Informasi / Berita Terkini / SARS-CoV-2
SARS-CoV-2
Introduction
Many countries are still struggling to control the COVID-19 pandemic, including Indonesia [1,2]. On December 31, 2020, Indonesia recorded 735,124 confirmed COVID-19 cases with 21,944 deaths and infection rate of approximately 7,000 cases/day [3].
One of the most important factors affecting the rapid spreading of COVID-19 is transmission within family [4,5]. It has been suggested the importance of the genomic epidemiology to fill the gaps in identifying the SARS-CoV-2 infection sources [6]. However, to our best knowledge, no reports described the genomic epidemiology within family clusters [6-8]. Moreover, multiple spike protein mutations have been associated with a higher transmissibility of SARS-CoV-2 [9]. In this study, we aimed to: 1) perform full genome characterization of SARS-CoV-2 and correlate the sequences with the epidemiological data within family clusters in Indonesia, and 2) conduct phylogenetic analysis of all samples from Yogyakarta and Central Java, Indonesia, involving the family cluster, and virus from other regions in Indonesia.
Methods
COVID-19 severity classifications
COVID-19 severity was determined based on the WHO classifications: 1) mild, without evidence of hypoxia or pneumonia; 2) moderate, pneumonia but not severe; 3) severe, pneumonia plus one of the following signs: respiratory rate >30 breaths/minute, severe respiratory distress, or SpO₂ <90% in room air; and 4) critical, acute respiratory distress syndrome (ARDS), sepsis, or septic shock, or other complications [10,11].
SARS-CoV-2 samples
We collected all virus samples of confirmed COVID-19 patients from Yogyakarta and Central Java provinces from June to November 2020. All nasopharyngeal samples were collected in viral transport media (DNA/RNA ShieldTM Collection Tube with Swab, Zymo Research, CA, United States) and transported into four COVID-19 diagnostic laboratories in Yogyakarta province: 1) Molecular Diagnostic Laboratory, Integrated Laboratory Unit, Dr. Sardjito Hospital; 2) Department of Microbiology and Laboratorium Diagnostik Yayasan Tahija World Mosquito Program, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada; 3) Balai Besar Teknik Kesehatan Lingkungan dan Pengendalian Penyakit (BBTKLPP), Yogyakarta; and 4) Disease Investigation Center, Wates, Yogyakarta. SARS-CoV-2 was detected by Real-Q 2019-nCoV Detection Kit (BioSewoom, Seoul, South Korea) with LightCycler® 480 Instrument II (Roche Diagnostics, Mannheim, Germany).
Full-genome sequencing
First, we performed RNA extraction of 19 nasopharyngeal swab samples by a QiAMP Viral RNA mini kit (Qiagen, Hilden, Germany), synthesized the double-stranded cDNA by Maxima H Minus Double-Stranded cDNA Synthesis (Thermo Fisher Scientific, MA, United States), and purified the cDNA using a GeneJET PCR Purification Kit (Thermo Fisher Scientific, MA, United States). For library preparations, we utilized the Nextera DNA Flex for Enrichment using Respiratory Virus Oligos Panel, whereas for full-genome sequencing, we used next generation sequencing (NGS) applied in the Illumina MiSeq instrument (Illumina, San Diego, CA, United States) with Illumina MiSeq reagents v3 150 cycles (2 x 75 cycles). We excluded two samples for further bioinformatics analysis because of low coverages. Our sample genomes were assembled by mapping to the reference genome from Wuhan, China (hCoV-19/Wuhan/Hu-1/2019, GenBank accession number: NC_045512.2) using Burrow-Wheeler Aligner (BWA) algorithm embedded in UGENE v. 1.30 [12]. Identification of single nucleotide polymorphism (SNP) was performed using the number of high confidence base calls (consensus sequence variations of the assembly) that disagree with the reference bases for the genome position of interest, then all SNPs were exported to a vcf file and visualized in MS Excel. The following accession IDs for 17 samples are: EPI_ISL_516800, EPI_ISL_516806, EPI_ISL_516829, EPI_ISL_525492, EPI_ISL_576383, EPI_ISL_632936, EPI_ISL_610161, EPI_ISL_610162, EPI_ISL_576145, EPI_ISL_632937, EPI_ISL_575331, EPI_ISL_576113, EPI_ISL_576114, EPI_ISL_576115, EPI_ISL_576116, EPI_ISL_576128, and EPI_ISL_576130 [13]. The first four IDs have been reported in our previous study [11].
Phylogenetic analysis
We used the reference genome of hCoV-19/Wuhan/Hu-1/2019 (NC_045512.2) for annotation of our sequences. A dataset of 142 available SARS-CoV-2 genomes (89 sequences from Indonesia and 53 from other countries) was retrieved from GISAID to conduct a phylogenetic analysis (Acknowledgment Table is provided in Supplementary Data). We only used the full-genome sequences of several strains representing SARS-CoV-2 clades from some countries that had complete genome and no long stretches of ‘NNNN’ for the phylogenetic analysis. MAFFT program server was utilized for multiple nucleotide sequence alignment (https://mafft.cbrc.jp/alignment/server/). A phylogenetic tree was constructed from 29.409 nt length of the open reading frame (ORF) of 142 SARS-CoV-2 virus sequences using Neighbor Joining statistical method with 2,000 bootstrap replications. The evolutionary distances were computed using the Kimura 2-parameter method and the rate variation among sites was modelled using a gamma distribution with estimated shape parameter (α) for the dataset. The estimation of α gamma distribution was calculated in DAMBE version 7 [14] , whereas all the other analyses was performed in MEGA version 10 (MEGA X) [15].
Ethical Approval
Our study was approved by Institutional Review Board of the Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada/Dr. Sardjito Hospital (KE/FK/0563/EC/2020). All participants or guardians signed a written informed consent for participating in this study.
Results
Severity and spike protein mutations of COVID-19 samples
Based on the case definition of COVID-19 severity developed for this study, 3 of 17 virus samples (17.6%) were collected each from asymptomatic case (people) and critical case, 5 virus samples (29.4%) from mild case, and 6 virus samples (35.3%) from moderate case (Table 1). Two of patients with critical stages eventually died. A range of Ct values found amongst different stage of severity, nevertheless all the virus samples with D614G mutations, except one (YO-UGM-10004/2020|EPI_ISL_576116), showed lower Ct values (clade GH, GR, and O, Ct range 16.9 to 24.7) than those with no mutation in this position (clade L, Ct 27.9).
Phylogenetic analysis
Phylogenetic analysis revealed that thirteen virus samples were situated within clade GH (GISAID classification), while two viruses were grouped with other viruses belonged to clade GR, and each one virus belonged to clade O and clade L (Figure 1). Three viruses from family cluster case-1 (YO-UGM-10001|EPI_ISL_576113, YO-UGM-10002|EPI_ISL_576114, and YO-UGM-10003|EPI_ISL_576115) formed a single group within clade GH, whereas viruses from cluster-2 (YO-UGM-1004|EPI_ISL_576116, YO-UGM-1005|EPI_ISL_576128, and YO-UGM-1006|EPI_ISL_576130,) were separated into two different nodes within clade GH (Figure 1, above).
Molecular analysis
Ninety-four SNPs were detected throughout open reading frame (ORF) of SARS-CoV-2 virus samples with 58% (54/94) nucleic acid changes resulted in amino acid mutations (Table 1, detailed in Table S.1). About 94% (16/17) virus samples showed D614G on spike protein and 56% of these (9/16) showed other amino acid mutations on this protein, including L5F, V83Ll V213A, W258R, Q677H, and N811I. Various amino acid mutations were also found in the other viral proteins, including on NSP2 (A205V, V247A, T256I, Q321K), NSP3 (P679S, P822L, T1022I, A1179V, T1198K, F1354C, P1665L), NSP4 (A231V), NSP5 (K12R, M49I, P184S), NSP6 (L37F), NSP8 (A21T), NSP9 (L42F), NSP12/RdRp (A97V, P227L, T248I, P323L, A656S, H892Y, M906V
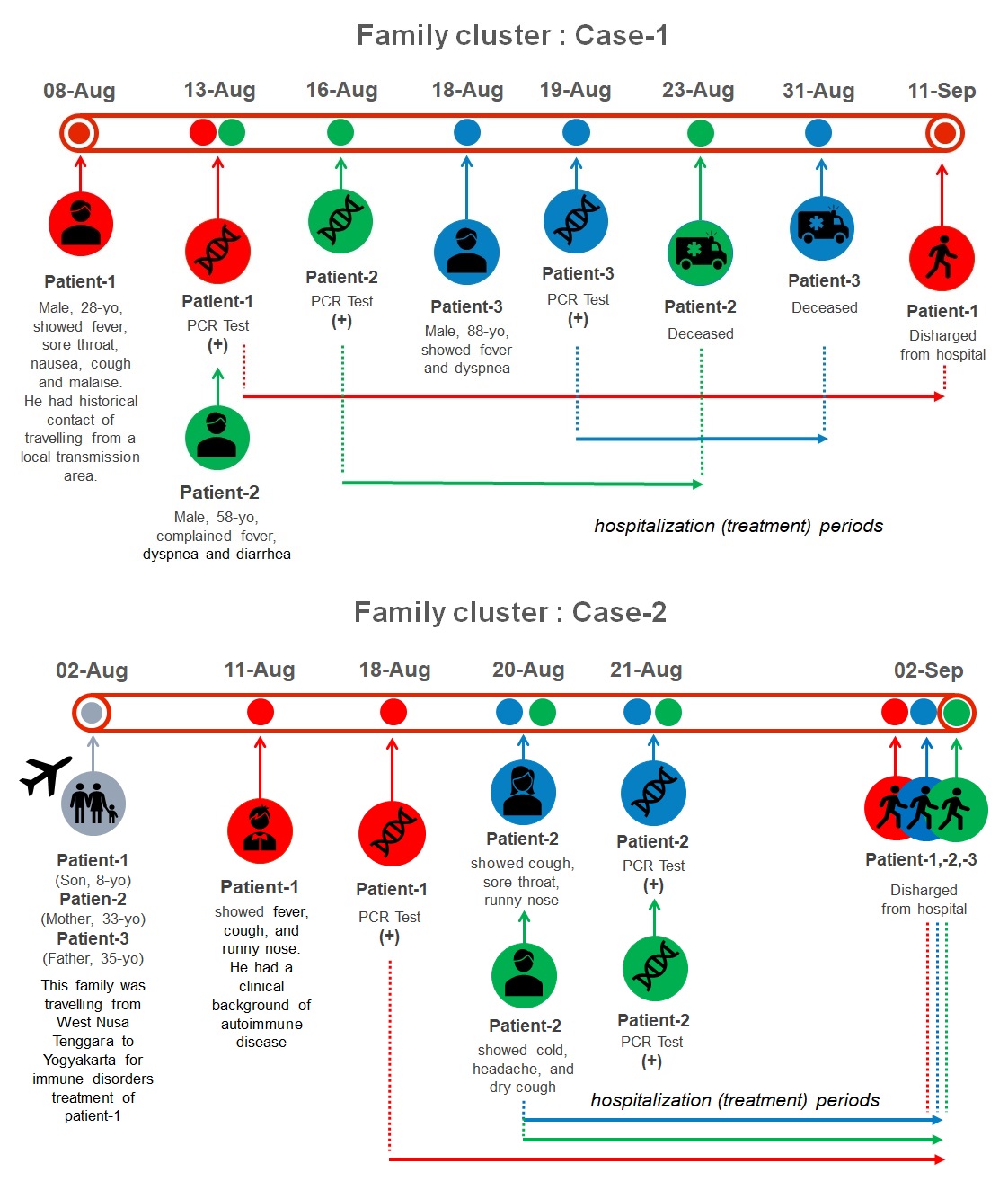
COVID-19’s family cluster: disease outcomes and molecular characterizations (Figure 2, Table 4)
Case presentations of COVID-19 patients among family cluster-1 and -2 are provided in Table 1. In family cluster-1, all three patients showed critical COVID-19 and eventually, two died (YO-UGM-10001|EPI_ISL_576113 and YO-UGM-10003|EPI_ISL_576115) and one survived (YO-UGM-10002|EPI_ISL_576114). In family cluster-2, the parents (EPI_ISL_576116, 576128) revealed mild COVID-19; while their son, an 8-year-old boy (EPI_ISL_576130), exhibited moderate COVID-19. All these family members were eventually survived (Table 1).
Analysis of multiple spike protein variants
All samples in the present study, except one, had D614G mutation. In addition, of 17 samples, 53% (9/17) showed other spike protein mutation besides D614G, including L5F, V213A, W258R, Q677H, and K811I (Table 2).
An interesting finding was documented from family cluster-1, in which all virus samples in this family cluster belong to the same clade GH, but two patients died and one survived (Table 2). All samples from family cluster-1 revealed other spike protein mutation, V213A, besides D614G. Samples from family cluster-2 (n=3) also belonged to clade GH and showed other spike protein mutation, i.e. L5F.
Moreover, the SNPs and amino acid variants detected in our 17 samples were summarized in Table 3 and 4, respectively (Reference sequence: NC_045512.2).
Discussion
Our study provides evidence of SARS-CoV-2 transmission within family, in which the same mutation of the spike protein in each family cluster was identified. It is important to understand the transmission routes of SARS-CoV-2 to prevent and control its spreading [4]. Families have been reported as the most dominant infection cluster of COVID-19 [16]. Family clusters have a higher risk of cross-infection because of frequent and close contact among each family member [4]. Our study also documented that although all family members showed the same multiple protein S mutations, however, they revealed the different outcomes. Additionally, another mutation in spike protein besides D614G was detected in all patients within family cluster 1, i.e. V213A.Whether this V213A mutation is associated with increased risk of mortality in COVID-19 patient requires further investigations.
Recently, more than 50% of the viral genome sequences in the UK was reported to have a new single phylogenetic cluster, i.e. VUI-202012/01 variant (multiple spike protein mutations: deletion 69-70, deletion 144, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H) [9]. These new variants have been associated with a higher transmissibility of SARS-CoV-2 up to 70% [9]. Until the submission date of December 2020 in GISAID, these variants were also detected in Asia [13]. Interestingly, we detected other spike protein mutations in our collected virus strains, including those from the family clusters, i.e. L5F, V213A, W258R, Q677H, and K811I. Noteworthy, V213A variant was identified in all patients from family cluster-1. V213A was detected in 4/17 (23.5%) of our samples. This variant is only found in 0.01% samples in four countries, including Indonesia [13]. Whether this variant is due to a founder effect, it needs a further study.
Currently, besides the D614G variant, several mutations within the receptor binding domains (RBD) of S protein have attracted most scientists’ attention due to their increased frequency in certain countries, including S477N (Australia and some Central European), N439K (UK and European), N501Y (part of the new UK variant VUI-202012/01 and the new South Africa variant 501.V2) [13]. These variants might be associated with some potential advantages for these viruses. However, whether these variants associated with COVID-19 clinical severity or SARS-CoV-2 transmission efficiency are poorly understood [17,18].
In addition, among eight clades in GISAID classification, we only detected five clades, i.e. L, G, GH, GR, and O, in SARS-CoV-2 samples from Indonesia and most of them (~60%) contained D614G. Globally, D614G has been detected in ~90% samples in 132 countries [13]. A recent study showed that D614G mutation is significantly associated with the increase of SARS-CoV-2 infectivity, competitive fitness, and transmission in primary human airway epithelial cells and hamsters [19].
Phylogenetic analysis showed that the full-genome sequences of SARS-CoV-2 identified within these family clusters are identical, strongly indicates a direct transmission within these families. Moreover, our study is also able to determine the virus clades of COVID-19 cases with unknown contact history with a confirmed COVID-19 cases. Our findings support a previous suggestion regarding the importance of genomic epidemiology in filling the gaps of identifying SARS-CoV-2 infection sources [6]. Therefore, a full-genome surveillance of SARS-CoV-2 in Indonesia is essential to prevent further transmission of SARS-CoV-2 and to identify a established or new variant that might affect the SARS-CoV-2 transmission.
Notably, our study only included a limited number of family clusters from Yogyakarta and Central Java, Indonesia. These limitations should be considered for interpretations of our findings.
Conclusions
Our study is the first comprehensive report associating the full-genome sequences of SARS-CoV-2 with the epidemiological data within family clusters. It highlights the same spike protein mutations among the same family member might show the different clinical outcomes. Moreover, we also detect multiple spike protein mutations in our samples. Further studies are necessary to clarify the impact of these multiple spike protein mutations in the transmission and severity of SARS-CoV-2 infection , especially in Indonesia.






